


■ 11.慢性心不全の心臓治療
11.1 連結橋の「エネルギー張力変換比率」とmyosin ATPase活性
一個の連結橋が回転して発生する張力(f)を単位時間に消費するエネルギー量(eA)で割ると,これは活性連結橋一個あたりの化学的エネルギーから機械的張力への「エネルギー張力変換比率(f/eA)」を表す。心筋のUm-FTI関係(tension dependent heatとforce-time-integralの関係)を実測し,その関係の傾き(eA/f)の逆数を計算すると連結橋の「エネルギー張力変換比率(f/eA)」が得られる(式7-5)。心筋の化学的エネルギーから機械的張力への変換はエネルギー源であるATPを加水分解するmyosin ATPaseによってなされる。myosin ATPase活性が低下すると「エネルギー張力変換比率」の値は増大するという関係にある。一個一個の連結橋の力が総合されて心筋の発生張力となり,最終的にはその心筋の短縮を通じて大動脈圧に逆らった左室のなす外的仕事に変換されてゆく。従って連結橋一個の「エネルギー張力変換比率」は心臓のエネルギー効率を確定する最も根本的なパラメーターの一つと考えられる。「エネルギー張力変換比率」の値は心不全においてはとりわけ重要である。なぜならこの値が高ければ同じ心筋張力を発生するのに少ないエネルギー消費ですみ,過負荷で疲弊している心筋を保護することに他ならないからである。
myosin heavy-chain isoformにはα-myosin(高いATPase活性)とβ-myosin(低いATPase活性)の二種類が存在し,ラットでは加齢(β優位),運動(α優位),圧負荷(β優位),甲状腺機能亢進(α優位),甲状腺機能低下(β優位),糖尿病(β優位),副腎不全(β優位)等で遺伝子発現を変換する92)。ヒトの心室筋はほとんど全てがβ-myosinから構成される。既に発表されたHasenfussらの摘出心筋でのデータ(文献71)のTables. 1と2,文献72)のFigs. 3と4)から心筋のUm-FTI関係の傾き(tension dependent heatとtension-time-integralの比)を計算し,その値から様々な摘出心筋における連結橋の「エネルギー張力変換比率」やmyosin ATPase活性についてこの理論モデルで解釈する。
摘出したヒト正常心筋と心不全心筋におけるUm-FTI関係の傾きは0.506と0.345mJ・mm2/mN・sec・gであり心不全心筋の方が低値であった(表3)。ヒトの正常心筋とvolume-overload心筋では0.097と0.054 mJ・mm2/mN・sec・gであり,ウサギの正常,pressure-overload,hyperthyroidの心筋ではそれぞれ0.139,0.055,0.239 mJ・mm2/mN・sec・gであり,ラットの正常とhypothyroidの心筋では0.305と0.155 mJ・mm2/mN・sec・gの値であった。この理論ではUm-FTI関係の傾きの逆数と連結橋の「エネルギー張力変換比率(f/eA)」とは等価であり(式7-5),Um-FTI関係の傾きとmyosin ATPase活性は比例関係にある。従って,連結橋の「エネルギー張力変換比率」はヒト心不全とvolume-overloadの心筋,ウサギのpressure overloadの心筋,ラットのhypothyroidの心筋ではすべて増大しており(myosinATPase活性は低下),ウサギのhyperthyroidの心筋では低下している(myosin ATPase活性は増大)と解釈される。pressure-overload,hypothyroid,hyperthyroidの心筋でのUm-FTI関係の傾きから予測したmyosin ATPase活性の結果は前述の実測データと同じである。ヒト不全心の乳頭筋では筋原線維のATPase活性は低下しているが,抽出したmyosin ATPase活性は変化していないとの報告があるので124, 133),ヒト心不全心筋では連結橋の「エネルギー張力変換比率」はmyosin ATPase活性だけでなく,何か別の因子からも調節を受けている可能性が示唆される。しかし,摘出心筋ではなくヒト心不全のin vivo心筋においてmyosin ATPaseが果たしてどの様な活性状態にあるかは全く不明である。
ラットではmyosin ATPase活性が温度3)や交感神経活動を介するcAMP79, 211)で調節され,ヒトの心不全でも同様にmyosin ATPase活性がcAMPで調節される69)。myosin ATPase活性はisoformの変換によってその値が変化するだけでなく,恐らくisoformの変換とは無関係に急性にもその活性が変化することが予想される。「エネルギー張力変換比率」は主にmyosin ATPase活性によって規定され,myosin ATPase活性は細胞内の二次メッセンジャーであるcAMPを介して調節され,cAMPはphosphodiesterase (PDE)と細胞膜内のadenylate cyclaseの活性によって二重に調節され,adenylate cyclase活性は心室筋細胞への交感神経活動などを介したグアニンヌクレオチド蛋白(GsとGi)によって調節されている(図11-1)。従って,心不全心筋において連結橋の「エネルギー張力変換比率」を高める最も簡単で確実な方法は心室筋への交感神経活動を低下することである。
ヒトの重症心不全では全身の交感神経活動の著明な亢進があるにもかかわらず,心筋β受容体のdown regulation18)やグアニンヌクレオチド蛋白,adenylate cyclase活性の変化等によってか心筋細胞内cAMPが逆に減少したり44),上述のように実験データの理論的解釈から連結橋の「エネルギー張力変換比率」も増大するなど,心筋にとってエネルギー効率の良い好ましい方向に調節されているようである。これは重症心不全では全身の交感神経活性を高めながらも,心臓では心筋収縮性維持を犠牲にしても連結橋の「エネルギー張力変換比率」を増大して心筋を保護しようとする生体の危機に対する適応現象かも知れない。この新しい解釈はそれらの細胞内異常現象が心不全の原因であるとする従来の説とは反対の立場をとる。
重症心不全のヒト拡張型心筋症の摘出心筋では,Ca2+ transientのピークからの減少過程中の収縮早期からCa2+ transientがゆっくりと再上昇する異常現象(hump)が観察される66)(図11-2)。これは筋小胞体のCa2+再取り込み異常によるもので,心不全心筋でのCa2+過負荷や拡張機能悪化の主要な原因の一つであると考えられている66)。しかし我々はこの現象を別の視点で解釈してみる。重症心不全心筋では残された機能心筋は収縮性を増大させ心臓ポンプ性能低下を代償しなければならない。そのため収縮開始直前に正常心筋よりも大量のCa2+を筋小胞体から放出するが(ピークCa2+ transientの増高66)),それでも足りずに収縮早期から再度細胞内[Ca2+]を増大させて心筋発生張力をさらに補強していると解釈する(Ca2+のbooster [Ca2+]を増大させて心筋発生張力をさらに補強していると解釈する(Ca2+のbooster効果)。このCa2+のbooster効果がcAMP非依存性であるならより好都合である。なぜなら連結橋「エネルギー張力変換比率」を悪化させずに心筋張力だけを増大できるからである。Ca2+のbooster効果によって心筋拡張機能は明らかに犠牲になるけれども,重症心不全心筋ではcAMPが減少し心筋収縮性維持を犠牲にするのと同様,この現象も危機的状態における心筋収縮性増大のために生体の選択した適応現象かも知れない。
このように従来から心不全悪化の原因と考えられているこれらの現象を,実は大量の障害心筋の存在する重症心不全への生体の重大な適応であると著者は解釈した。そして,そこに慢性心不全に対する治療の基本原則の鍵が見られると推理し,以下に三つの心臓治療仮説を提案した。
11.2 慢性心不全に対する心臓治療仮説
急性投与では心筋収縮性増大や血行動態の改善が見られるcAMP依存性の強心剤(β受容体刺激剤やphosphodiesterase (PDE) 阻害剤)を長期間投与すると,プラセボと比較して死亡率が有意に悪化した60, 131, 195, 200)。この事実から慢性心不全に対する治療目標は,確実で明らかな心筋収縮性の増大や血行動態の改善ではなくて,六ヶ月以上にわたる長期的な観察に基づく症状と生命予後の改善であるとの考えが最近合意を得つつある。しかし,長期的な症状改善や生命予後の向上を実現するための具体的な治療戦略の基本原則が未だ不明である。
近年,慢性心不全治療について様々な種類の薬剤による効果が多数報告されている。その中で検討症例数がとりわけ多いangiotensin converting enzyme (ACE)阻害剤の慢性心不全の予後に対する有効性が注目されている136, 191, 193, 194, 217)。それほどの大規模研究ではないけれどもヒドララジンとISDNによる血管拡張療法25, 26)や拡張型心筋症に対するβ遮断剤202, 203)の有用性はほぼ確実であろう130)。右室への人工ペースメーカー植込み治療78),強心配糖体のジギタリス31, 56, 132,215),ある種の少量のPDE阻害剤43),Ca2+感受性増強剤98, 145)などが慢性心不全に有用かも知れないとの示唆的な報告もある。他方,末梢動脈拡張作用による後負荷減少,心不全心筋におけるCa過負荷の改善,虚血性心疾患における冠動脈血流増加作用等を期待されるはずのCa拮抗剤治療が虚血性心疾患を合併する心不全の死亡率と心不全発症のリスクを高め,心不全治療薬として必ずしも適切でないことが明らかになりつつある10, 37, 38, 48, 61, 192)。α受容体遮断剤は短期的には血行動態的に良好な効果があるが,長期投与では心不全症状や生存率に有利ではない25)。この理由はrenin-angiotensin系の賦活化に関係するらしいと考えられている11)。
ここで慢性心不全に対する心臓治療の基本原則に関する仮説を模索してみたい。ただし心不全の予後に影響を与えるが,心筋の力学的,エネルギー的問題と直接に関係のない不整脈・突然死や多くの体液因子の問題はここでは取り上げない。また心不全の原因が明らかであり,その治療法も確立しているものは除く。我々が推理した慢性心不全の心臓治療仮説とは,
仮説1):連結橋の「エネルギー張力変換比率(f/eA)」を高める
仮説2):心筋収縮性を適度に増大する
仮説3):減負荷療法によって心臓の外的仕事量を軽減する
である。Ca2+のトロポニンCへの親和性(モデルではα)を増大させて,心筋収縮に関与しないCa2+の処理エネルギーを減らす治療は効果が少ないと予測する。なぜならαは正常心筋で0.95 (上限は1.0)169),心不全では少し低下して0.92程度であり(5章7節参照),心不全でのαの増加効果は僅かしか期待できないとモデルから予想されるからである。
11.3 慢性不全心の薬剤治療仮説
以上の慢性心不全における心臓治療仮説から現在臨床の場で使用可能な医用器材や薬剤の範囲内で次の様な具体的な心臓治療方法を想定する。
仮説1):myosin ATPase活性を確実に低下させるために,β受容体遮断剤や右室への人工ペースメーカー植込み97)によって交感神経のβ受容体刺激を抑制する。しかしこの治療だけでは心筋の[Ca2+],発生張力,Kaも全て同時に低下することになるので,β受容体遮断剤投与によって心不全が悪化するリスクが高まる。人工ペースメーカー植込みでは交感神経活動の抑制は生理的反応として徐々に起こるので,心不全が急速に悪化する例はβ受容体遮断剤よりは少ないであろう。しかしリスクが無くなるわけではない184)。この治療だけでは心不全悪化のリスクを減らすために,心不全を悪化させるリスクが増えるという自己矛盾が生ずる。従って,この治療だけの心不全治療効果は,心不全悪化のリスク減少効果とリスク増大効果の
差によって決まる。
仮説2):β受容体刺激の抑制によって低下した心筋収縮性を適度なレベルまで回復,増加させる。心筋の収縮性を増大させるにはEcを増加させる。Ecは細胞内[Ca2+],Ca2+のトロポニンCへの親和性(α),単位活性連結橋の発生張力(f)三者の積である(式3-7)。単位活性連結橋の発生張力は連結橋の回転速度を決定するmyosin ATPase活性に比例するであろう。心筋の短縮速度はCa2+とトロポニンCとの反応速度定数であるKaとEcの積に比例する(式6-1)。従って,心筋の収縮性を亢進するには細胞内[Ca2+],Ca2+のトロポニンCへの親和性(α),myosin ATPase活性,Ca2+とトロポニンCとの反応速度定数(Ka)を増大させることである。cAMPは少なくとも[Ca2+],myosin ATPase活性,Ca2+とトロポニンCとの反応速度定数(Ka)を調節している。cAMPの増大はmyosin ATPase活性を高め「エネルギー張力変換比率」の低下につながるので,心筋収縮性を亢進させるためにはcAMP非依存性に陽性変力作用を示す薬剤が良い。細胞膜のNa+, Ka+-ATPase活性を抑制することによって細胞内[Ca2+]を増加させ36),同時にKaも増大21, 49, 208)させるジギタリスがこの条件に合致する薬剤の一つである。慢性心不全では既に大量の無機能障害心筋が存在するため,残余の機能心筋は収縮性の予備をかなり使用しているであろう66, 117)。この状態の残余機能心筋に過度の収縮張力増大を期待してはならない。心筋収縮性増大の治療は控えめにする方が良いであろう。
仮説3):心臓の外的仕事量を軽減するには,主要臓器への血流量と潅流圧の必要レベルを保つ限界近くまで大動脈圧と静脈環流を低下させ,左室サイズを縮小することである。以上の仮説1)と2)の治療が実現し,前負荷,後負荷を下げる余裕があれば,さらに左室の負荷を減少させることは有意義である。これによって心臓の全仕事量は減少し,左室拡張末期圧や肺毛細管圧も低下するであろう。ここでは交感神経は遮断されているので反射性の交感神経亢進は問題とはならない。この減負荷治療のために利尿剤,ヒドララジン,ISDNの使用が勧められる。ほとんどのCa拮抗剤は仮説2)の心筋収縮性増大に対し逆効果であり,特に重症心不全ではCa拮抗剤による減負荷治療は心不全悪化リスクを大きく増加させる。
以上の三つの治療を同時に適切に施すことによって,心筋収縮性を適度に増大した状態で,連結橋の「エネルギー張力変換効率」は向上し,左室のUm-Fp関係,Um-PVA関係,Um-FTI関係全ての傾きは低下し,さらに心血行動態を改善しつつ心臓の全外的仕事量の減少が達成できるかも知れない。もしこれらが全て実現すれば残された左室機能心筋の重い負担は軽減し,心臓全体のエネルギー効率が改善し,患者のquality of lifeは向上し,心不全発生率や死亡率は減少することが期待される。
11.4 ジギタリスとACE阻害剤の併用
無症状の軽症心不全にACE阻害剤を投与すると心不全の発症に予防効果があった194)。中等度の心不全にジギタリスまたはACE阻害剤を投与すると,プラセボと較べ両薬剤とも心不全の発生頻度が有意に減少した190)。利尿剤とジギタリスが既に投与されている中程度以上の心不全にACE阻害剤を追加投与すると,死亡率や心不全の発症率が有意に減少した191, 193)。angiotensin IIは交感神経末端や副腎髄質からのnoradrenaline遊出を増加するので,ACE阻害剤は治療仮説1)と3)に対する効果が期待される。ジギタリスには交感神経活動を抑制する直接作用があるので46, 47, 55),仮説1)と2)の治療効果が期待できる。利尿剤,ジギタリス,ACE阻害剤の三者を併用投与すれば,仮説1)にはジギタリスとACE阻害剤,仮説2)にはジギタリス,仮説3)に対してはACE阻害剤と利尿剤による治療が施されたことになる。以上の三者併用は完全ではないが慢性心不全に対する心臓治療仮説の意図に合致する。
11.5 PDE阻害剤,ジギタリス,ACE阻害剤の併用
利尿剤,ジギタリス,ACE阻害剤等を投与中の中等度以上の心不全患者にβ受容体刺激剤のxamoterol195)や純粋なPDE阻害剤であるenoximone,imazodan,milrinoneを追加投与すると死亡率がプラセボより有意に増大した60, 131, 200)。同様の治療を受けている中等度以上の心不全患者に別のPDE阻害剤であるvesnarinone (60 mg/day)を投与すると,左室駆出率は改善しなかったが死亡率や心不全の発症率がプラセボより有意に減少した43)。しかしこの患者に倍量のvesnarinone (120 mg/day)を投与すると死亡率がプラセボより有意に高くなった。この治療では仮説1)にはジギタリスとACE阻害剤,仮説2)にはジギタリスとvesnarinone,仮説3)に対してはACE阻害剤,利尿剤,vesnarinoneによる治療vesnarinone,仮説3)に対してはACE阻害剤,利尿剤,vesnarinoneによる治療が施されたことになる。PDE阻害剤はcAMPを増加させるので治療仮説1)と対立する。しかしvesnarinoneにはPDE阻害作用の他にもcAMP非依存性の陽性変力作用(細胞内へNa+の流入を増大させてCa2+の供給を増加させる)がある130)。60mg/dayの投与量では[Ca2+]の増大効果に比較しcAMPの増大は軽度であった可能性がある。60 mg/dayのvesnarinoneによるmyosin ATPase活性の亢進は,ジギタリスとACE阻害剤によるmyosin ATPase活性の低下を打ち消すほど強いものではなかったのかも知れない。一方,120 mg/dayのvesnarinoneによるmyosin ATPase活性の亢進は,ジギタリスとACE阻害剤によるmyosin ATPase活性の低下を相殺するほど強いものであったかのかも知れない。
11.6 Ca感受性増強薬
pimobendanは著明なアクチンフィラメントのCa 2+感受性増強作用といくらかのPDE阻害作用を持つ薬剤であると言われている。pimobendanがアクチンフィラメントのCa2+感受性を増強する薬剤であるとする主要な根拠は,同一の細胞内[Ca2+]条件でありながらpimobendan投与によって心筋の発生張力が増大するからである。しかしこの現象だけなら,1)Ca2+感受性増強作用以外にも,2)myosin ATPase活性の増大,3)活性連結橋とアクチンフィラメントとの結合時間の延長,4)トロポニンCに対してCa2+と同じ効果を持つCa2+ analogueによっても同様な結果がもたらされる可能性がある。
pimobendanは同じ細胞内[Ca2+]条件でCa2+と結合したトロポニンの濃度を増加させる作用を持つ薬剤(その増加程度は僅かでありpimobendanの強い陽性変力作用を説明するには充分とは思えないけれども,本来の正確な意味でのCa2+感受性増強作用である)であると報告52)した同じグループが,pimobendanの体内での活性代謝物であるUD-CG212 ClにはCa2+と結合したトロポニンの濃度を増加させる作用が認められないと報告した209)。従って,同一の細胞内[Ca2+]条件で心筋発生張力は増大するものの,UD-CG212 Clには本来の意味でのCa2+感受性増強作用は無いことになる。
pimobendanはmyosin ATPase活性に影響を与えず,この薬剤の心筋張力増加作用は活性連結橋とアクチンフィラメントとの結合時間の延長によってもたらされたものであると推測することもできる。もしそうであれば同一の[Ca2+]と消費エネルギーで連結橋はより大きな張力を出すことになるので,myosin ATPase活性は変化せずに「エネルギー張力変換効率」は向上することになる。しかし,この可能性は以下の実験で確実に否定される。
ラット心筋での心筋酸素消費量とstress-time-integral関係(U-FTI関係と等価)の傾きはpimobendanと対照で差が無かった70)。ヒトDCMの心不全におけるin vivo心筋酸素消費量とstress-time-integral関係の傾きでは,pimobendanとnitroprussideの値に差は無かった68)。同様の解析を摘出心筋においてisoproterenol,E1020(PDE阻害剤),ouabain,calcium, pimobendanで比較すると,isoproterenolやE1020での傾きに較べ,ouabain,calcium,pimobendanでの傾きはほぼ同様な程度に低値であった88)。これらの一連の心筋酸素消費量とstress-time-integral関係の傾きの研究結果からこの理論モデルを用いて推測した結論は,pimobendanは正常心筋と心不全心筋の両者においてmyosin ATPase活性や連結橋「エネルギー張力変換効率」にはなんら影響を与えず,あたかもouabainやcalciumがcAMP非依存性に細胞内[Ca2+]を増大させたのと同様な陽性変力作用とエネルギー消費動態を,細胞内[Ca2+]を変えずに実現することである。これを例えれば,[Ca2+]は1μMしか筋小胞体から放出されていないのに,pimobendanにはあたかも3μM放出したかのように心筋細胞を機能させる作用があることである。
pimobendanは同じ細胞内[Ca2+]条件であってもCa2+と結合したトロポニンCの濃度を増加させることによって心筋発生張力を増大させる作用を示すCa 2+感受性増強剤というよりも,それ自体がトロポニンCに対してCa2+と類似の効果を持つCa2+ analogueであると考えれば以上の報告は了解可能である。いずれにせよpimobendanやその代謝活性物であるUD-CG212 ClにはcAMP非依存性の陽性変力作用があることは確からしい。
11.7 Ca感受性増強薬,ジギタリス,ACE阻害剤の併用
利尿剤,ジギタリス,ACE阻害剤で治療されている中等度以上の心不全にpimobendanを三ヶ月間投与すると,pimobendan投与群では運動時間,最大酸素消費量が増加し,quality of lifeもプラセボに比較し有意に改善した98)。興味深いのはpimobendanの5 mg/dayの投与量が2.5や10 mg/dayよりもいずれの指標でも有効性が最も高かったことである。5 mg/dayより多くても少なくても心不全に対する効果は低下した。左室駆出率,血中catecholamine濃度,催不整脈作用ではプラセボ群と実薬群間に差はなかった。心不全の悪化リスクは実薬群で有意に低かったが死亡率には差がなかった98)。利尿剤,ジギタリス,ACE阻害剤,pimobendanの併用投与は中程度以上の慢性心不全において心不全悪化リスクを減少させ活動能力を改善した98, 145)。この治療では仮説1)にはジギタリスとACE阻害剤,仮説2)にはジギタリスとpimobendan,仮説3)に対してはACE阻害剤,利尿剤,pimobendanによる治療が施されたことになる。左室駆出率に差はなかったのでこの投与量では陽性変力作用は恐らく軽度であろう。
pimobendanの体内での活性代謝物であるUD-CG212 Clの示す陽性変力作用は主にcAMP依存性である可能性も指摘されている39)。pimobendanは5 mg/dayの投与量がcAMP増加が相対的に少なく,cAMP非依存性の陽性変力効果がある程度期待できる至適量なのかも知れない。10 mg/dayではcAMP増加による悪影響が出現し,2.5 mg/dayでは陽性変力効果が不足なのかも知れない。pimobendanの長期投与の有効性は、vesnarinoneと同様に,活性代謝物であるUD-CG212 Cl のcAMP増加作用とcAMP非依存性陽性変力作用のどちらがより優位であるかによって決定するのであろうか?。それはpimobendanの六ヶ月以上にわたる長期投与の大規模試験の結果で明らかになるかも知れない。
11.8 心臓治療仮説の優先順位
確実な心筋収縮性亢進や心血行動態的改善を得るためにcAMPを増加させる純粋なPDE阻害剤やcatecholamineを投与すると,心不全の予後がかえって悪化してしまうのは,myosin ATPase活性増加による連結橋の「エネルギー張力変換効率」低下と[Ca2+]の増加を含む強力な陽性変力作用が原因の一つと思われる。しかしcAMP非依存性の陽性変力作用を示すジギタリスとvesnarinoneでは心不全治療に良い結果が得られたことから,cAMP増加による「エネルギー張力変換効率」低下の方が陽性変力効果よりも心不全悪化に対する影響が大きいことが示唆される。利尿剤,ACE阻害剤,ジギタリス,少量のvesnarinoneの併用で左室駆出率が変化しない程度の弱い心筋収縮性の増加であっても心不全の予後が改善されたことから,仮説2)の心筋収縮性増大は軽度にとどめることの重要性が示唆される。また利尿剤とジギタリスが投与された心不全にヒドララジンとISDNまたはACE阻害剤を追加した時,ACE阻害剤を追加した群の方が血行動態的効果で劣っていたにもかかわらず死亡率が有意に低かったことは26),仮説3)の血行動態的に有効である減負荷療法よりも仮説1)の「エネルギー張力変換効率」向上の治療がより重要であることを示唆する。従って慢性心不全の治療には「エネルギー張力変換効率」を向上する仮説1)が他の二つの仮説より優先度が高く,まず最初になされるべき治療であると思われる。
結論として,
一見共通性のないジギタリス,β遮断剤,ACE阻害剤,長期の右室人工ペーシング治療は、いずれも主に連結橋の「エネルギー張力変換比率」を向上するが故に慢性心不全治療として有効であり,短期的にはいずれも明らかに強力な強心剤であるxamoterol,enoximone,imazodan,milrinone,vesnarinone(120 mg/day)は「エネルギー張力変換比率」を低下させる故に慢性心不全の予後を悪化すると著者は推測した。
11.9 心臓治療仮説の限界
心不全の予後に影響を与える可能性のあるその他の問題として左室心筋虚血,左室拡張障害,左室間質組織障害,左室のremodeling,不整脈・突然死,電解質異常,体液因子(renin-angiotensin,vasopressin,endotherin,endotherium derived relaxing factor ,cytokines,free radical,nitric oxide),心筋細胞膜β受容体のdown regulation,グアニンヌクレオチド蛋白(GsとGi)やadenylate cyclaseの変化,心筋内Ca2+過負荷,心筋内で産生される体液因子(ANP,BNP,angiotensin),ACE阻害作用以外のACE阻害剤自体の何らかの心筋への直接作用125)などがある。ここではこれらの多くの因子について検討を加えていない。以上の種々の因子については,心不全の病態・治療との関連について今後更に詳細な実験的,臨床的知見が得られることを期待する。
心不全とは極めて複雑で多彩な因子から構成される全身的症候群であるが,ここで提示した心不全における心臓治療仮説は,その極く一部の因子だけを対象として想定されたものに過ぎないことを強調する。
表11-1:様々な心筋における単位連結橋のエネルギー張力変換比率(f/eA)
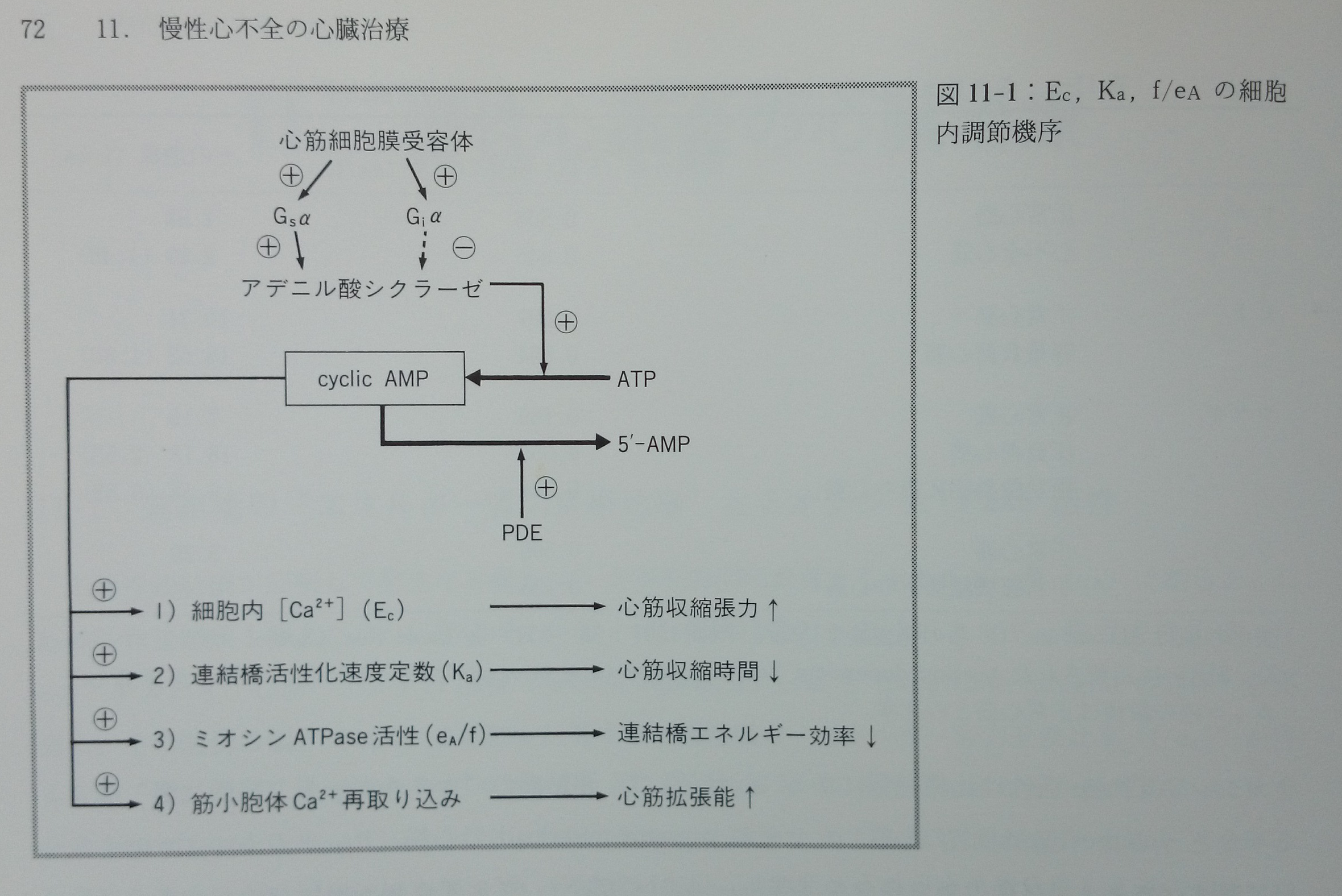
図11-1:Ec,Ka,f/eAの細胞内調節機序。
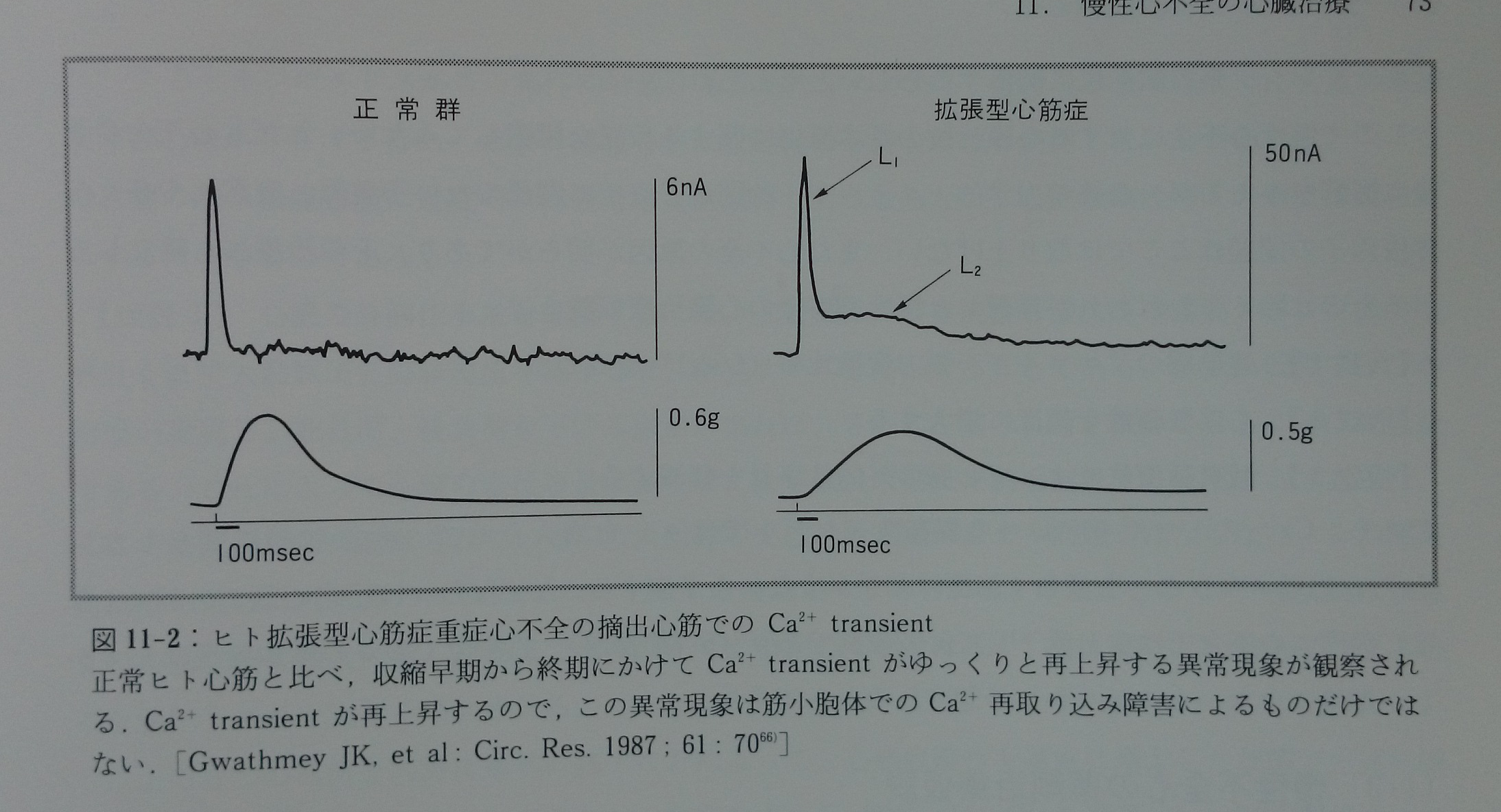
図11-2:ヒト拡張型心筋症重症心不全の摘出心筋でのCa2+ transient。正常ヒト心筋と較べ,収縮早期から終期にかけてCa2+ transientがゆっくりと再上昇する異常現象が観察される。Ca2+ transientが再上昇するので,この異常現象は筋小胞体での再取り込み障害によるものではない。[Gwathmey JK, et al: Circ. Res.1987;61:7066)]
★心不全治療の原理をミステリー風に解説

慢性心不全治療の原理理論を、一般の読者にも理解し易いよう推理読み物にまとめました。ミステリースタイルの要約です。